生物信息学分析
- 产品详情
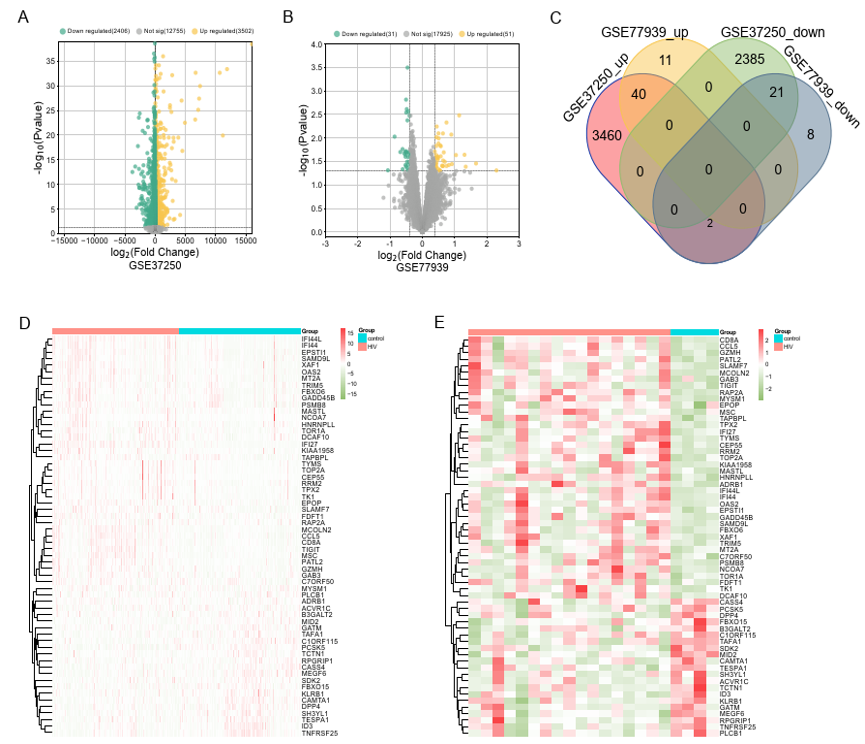
1. 差异性表达基因筛选(DEGs)
从GEO数据库下载。利用 R 软件的 Limma 包来识别 DEGs。将阈值设置为“调整 P 值 < 0.05 和 |logFC|> 0.38“ 用于识别 DEGs。
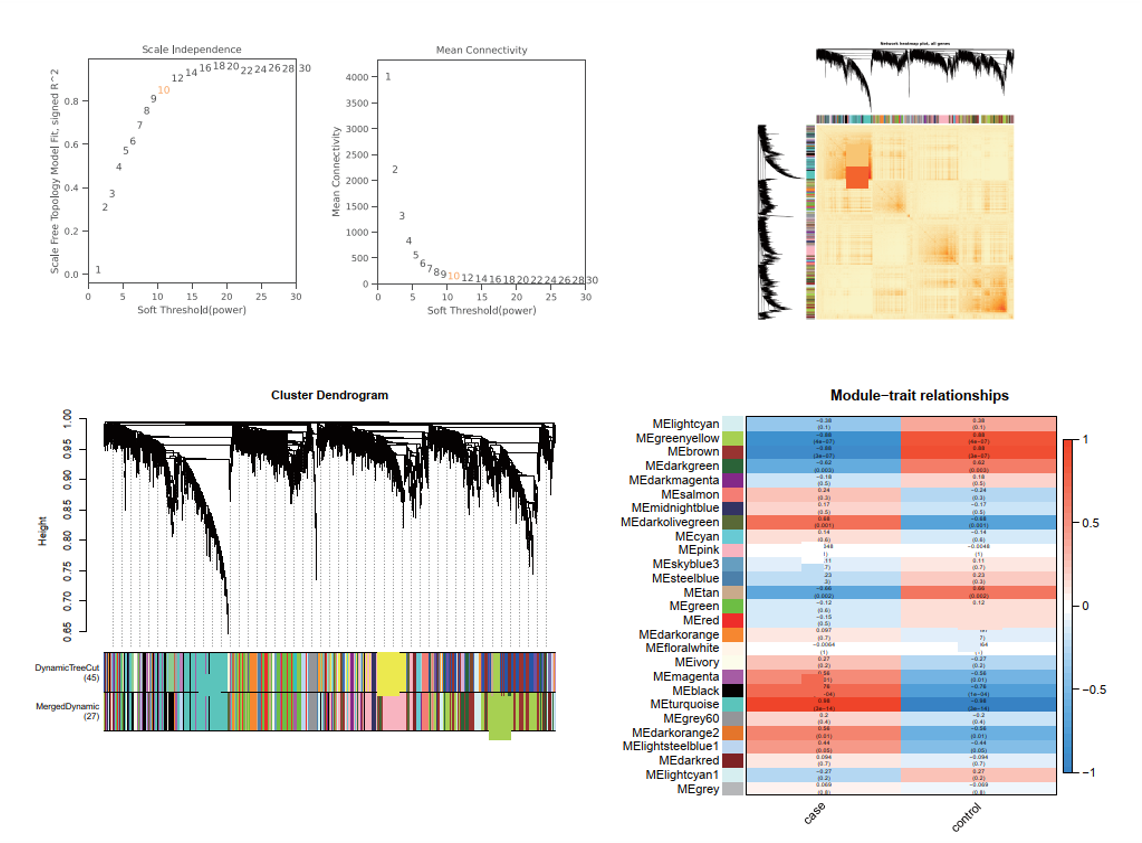
2. WGCNA分析
使用R软件通过WGCNA方法鉴定与疾病高度相关的基因簇。使用 pickSoftThreshold R 函数确定软阈值功效 (β),基于加权共表达式网络对彩色模块进行分层聚类。
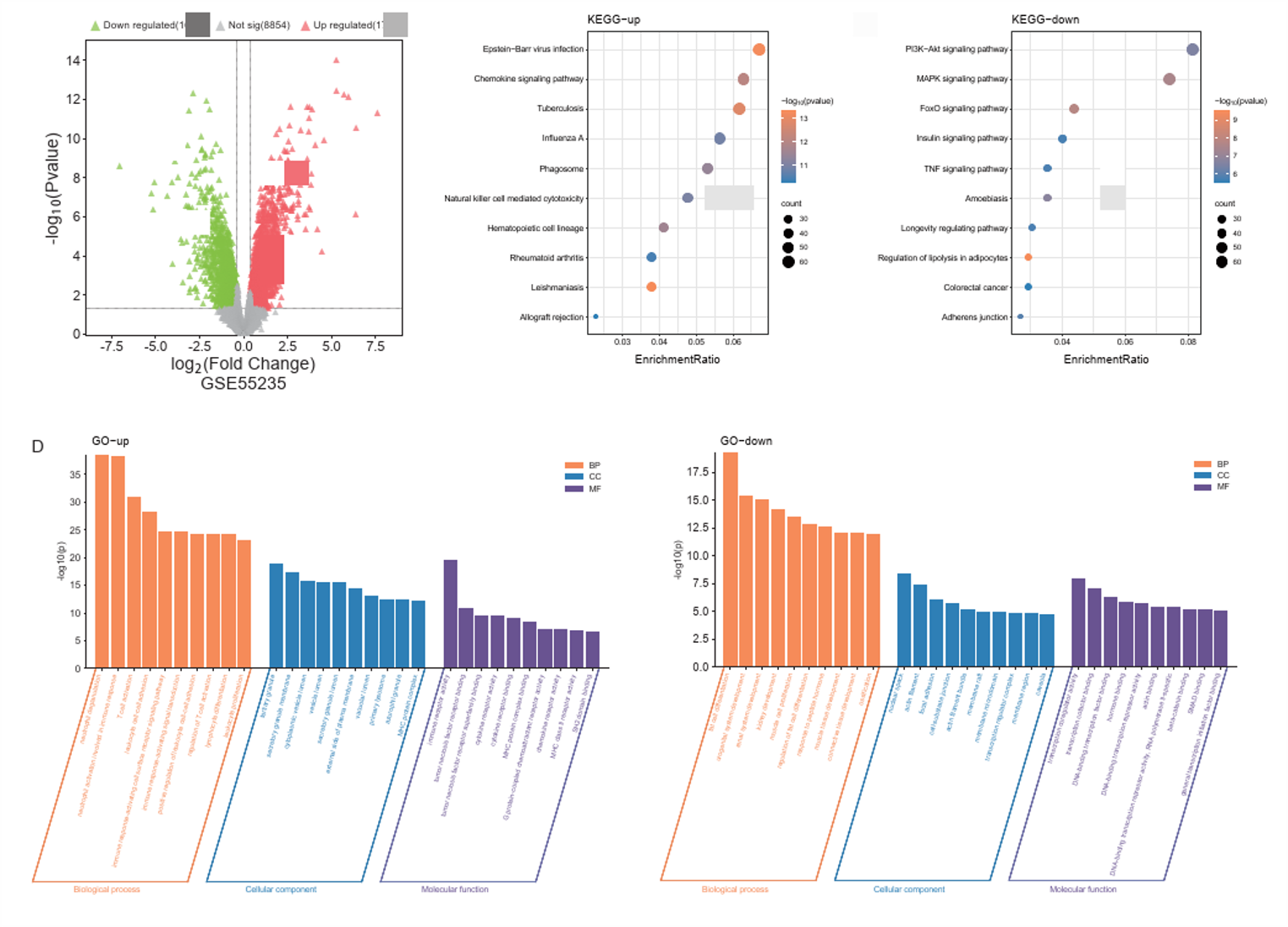
3. KEGG分析和GO分析
利用R包进行基因本体(GO)功能富集分析和京都基因与基因组百科全书(KEGG)通路富集分析。GO分析主要用于注释基因功能,特别是生物通路(BP)、细胞成分(CC)和分子功能(MF)。采用KEGG分析检测DEGs中的通路富集情况。
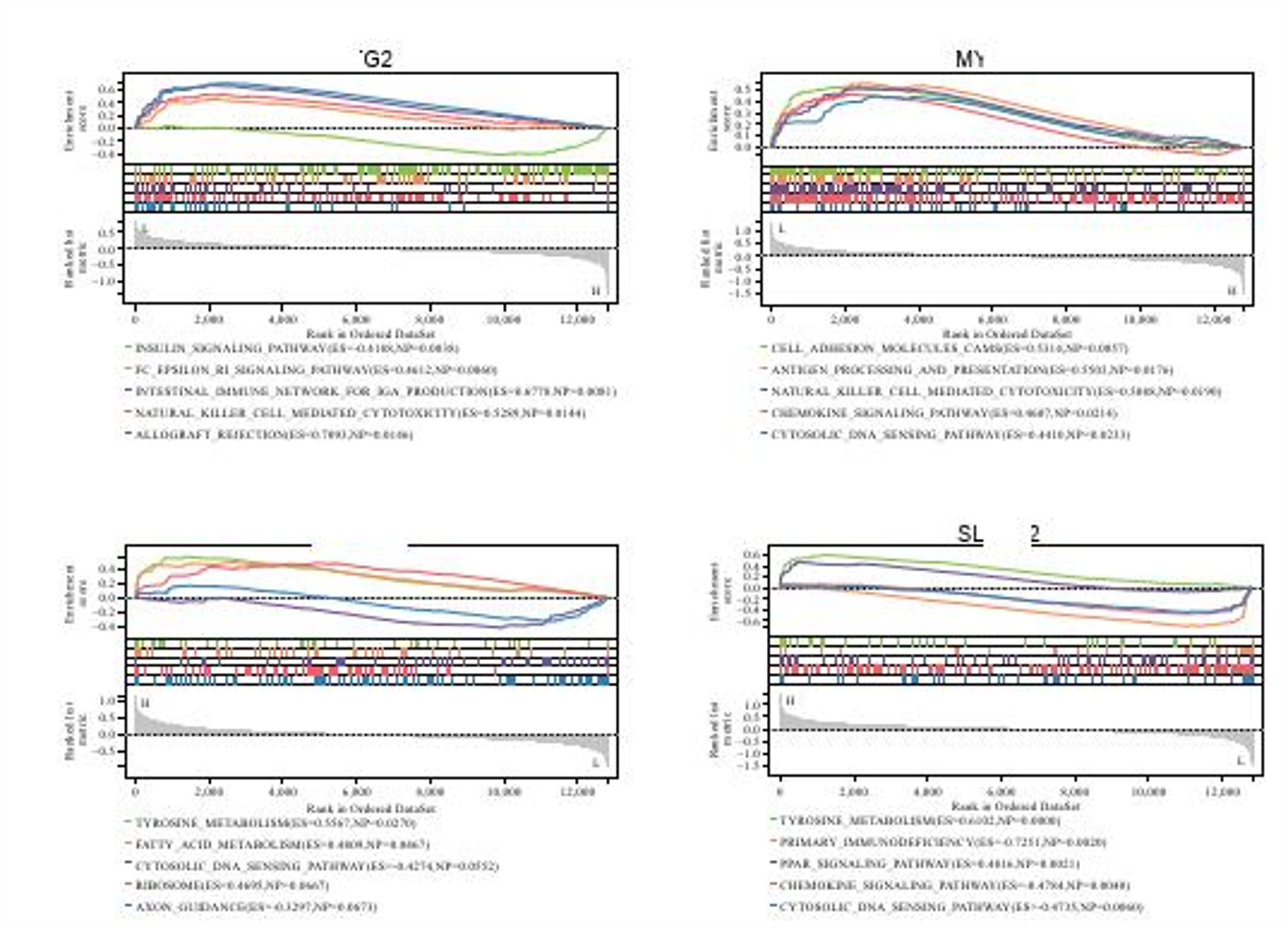
4. GSEA分析
利用GSEA软件(version 3.0),根据基因表达水平将样本分成高表达组(>=50%)和低表达组(<50%),并从Molecular Signatures Database)下载了c2.cp.kegg.v7.4.symbols.gmt子集合,用以评估相关途径和分子机制,基于基因表达谱和表型分组。
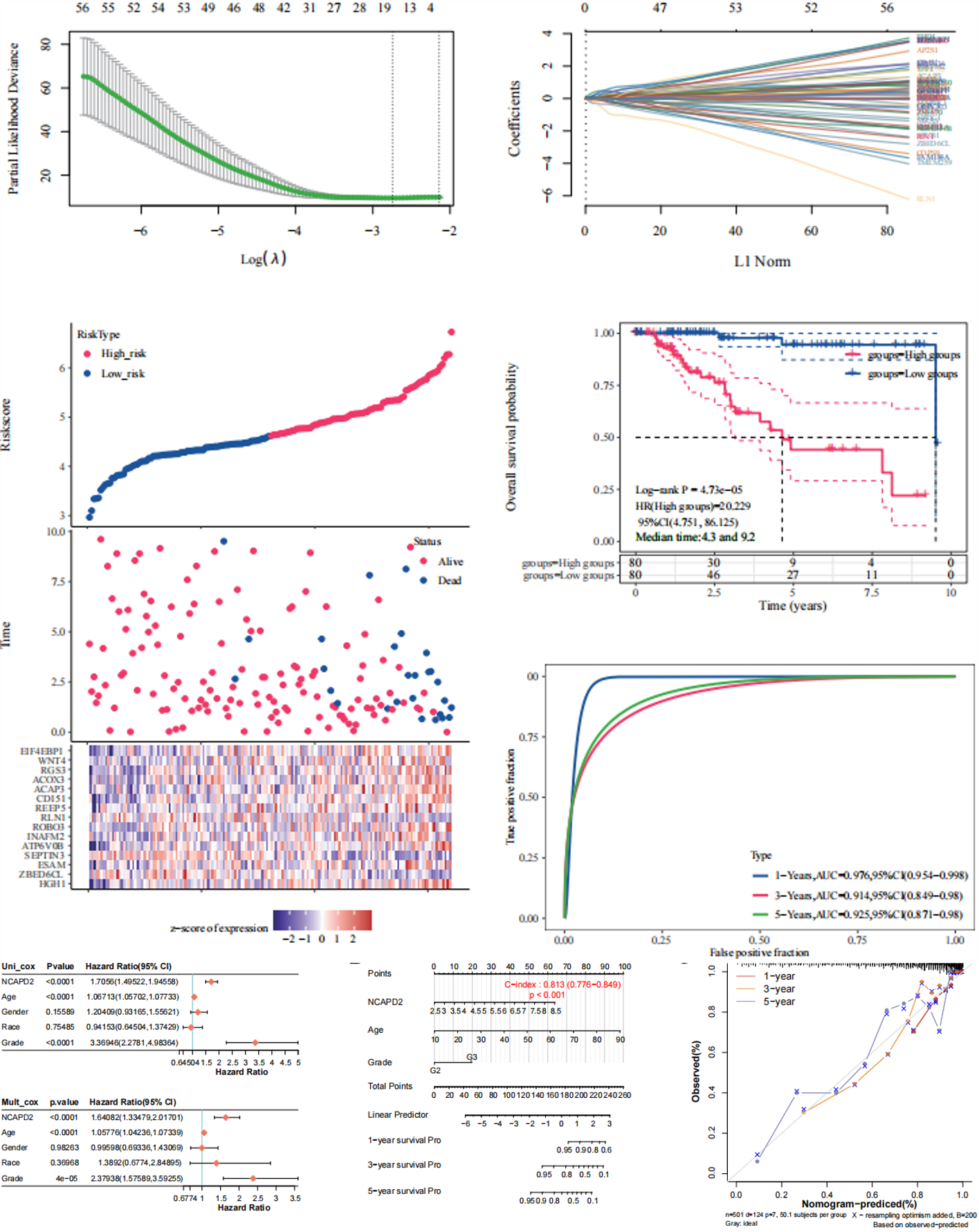
5. 肿瘤预后模型构建
Lasso cox回归选定特征的系数由lambda参数显示,横坐标代表自变量 lambda 的值,纵坐标表示自变量的系数;使用LASSO Cox回归模型绘制了部分似然偏差与log(λ)的关系。
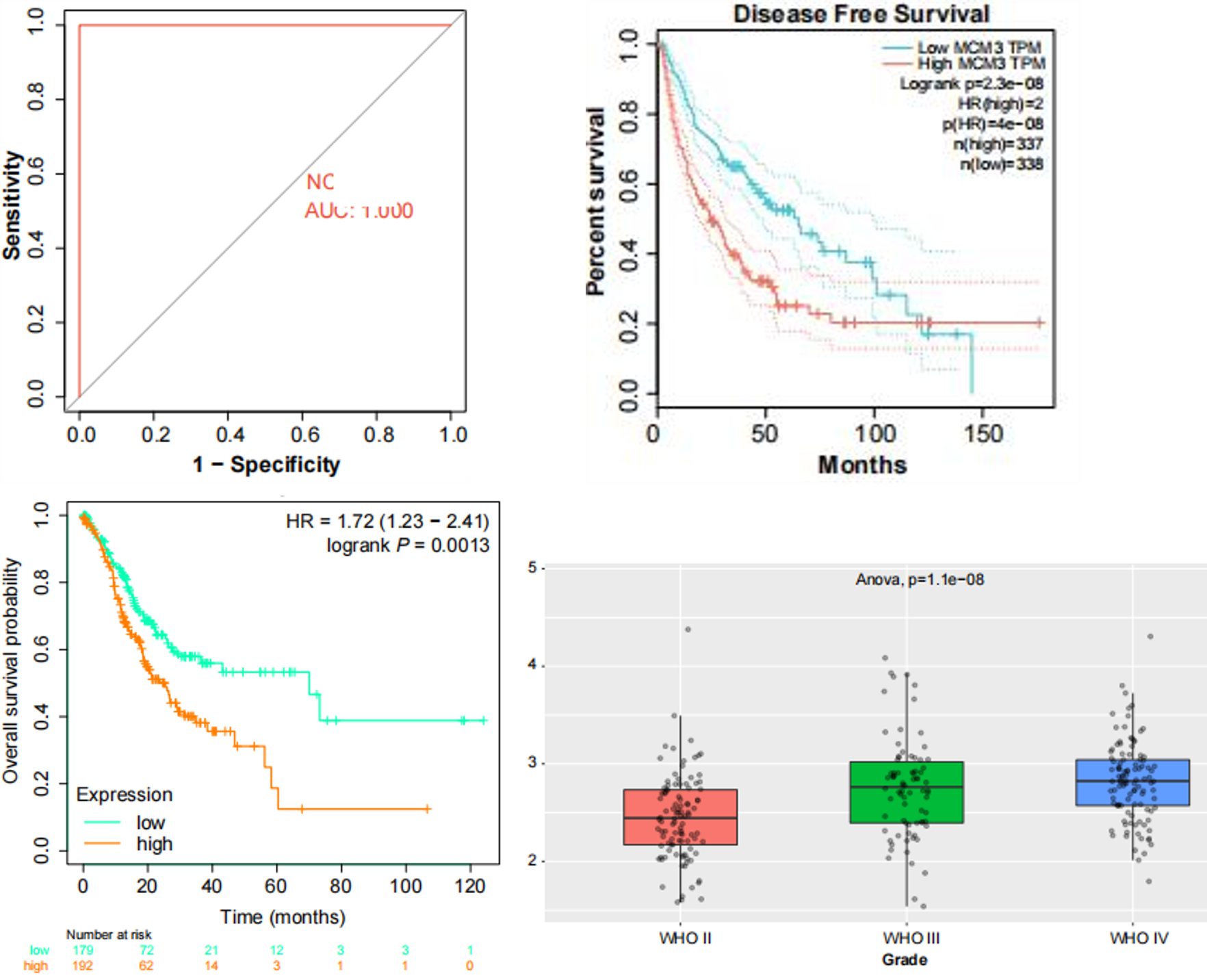
6. 临床相关分析
从数据库中获得肿瘤的RNAseq数据和相应的临床信息。log rank用于检验KM生存分析比较上述两组或多组之间的生存差异,进行了timeROC分析以比较基因的预测准确性。
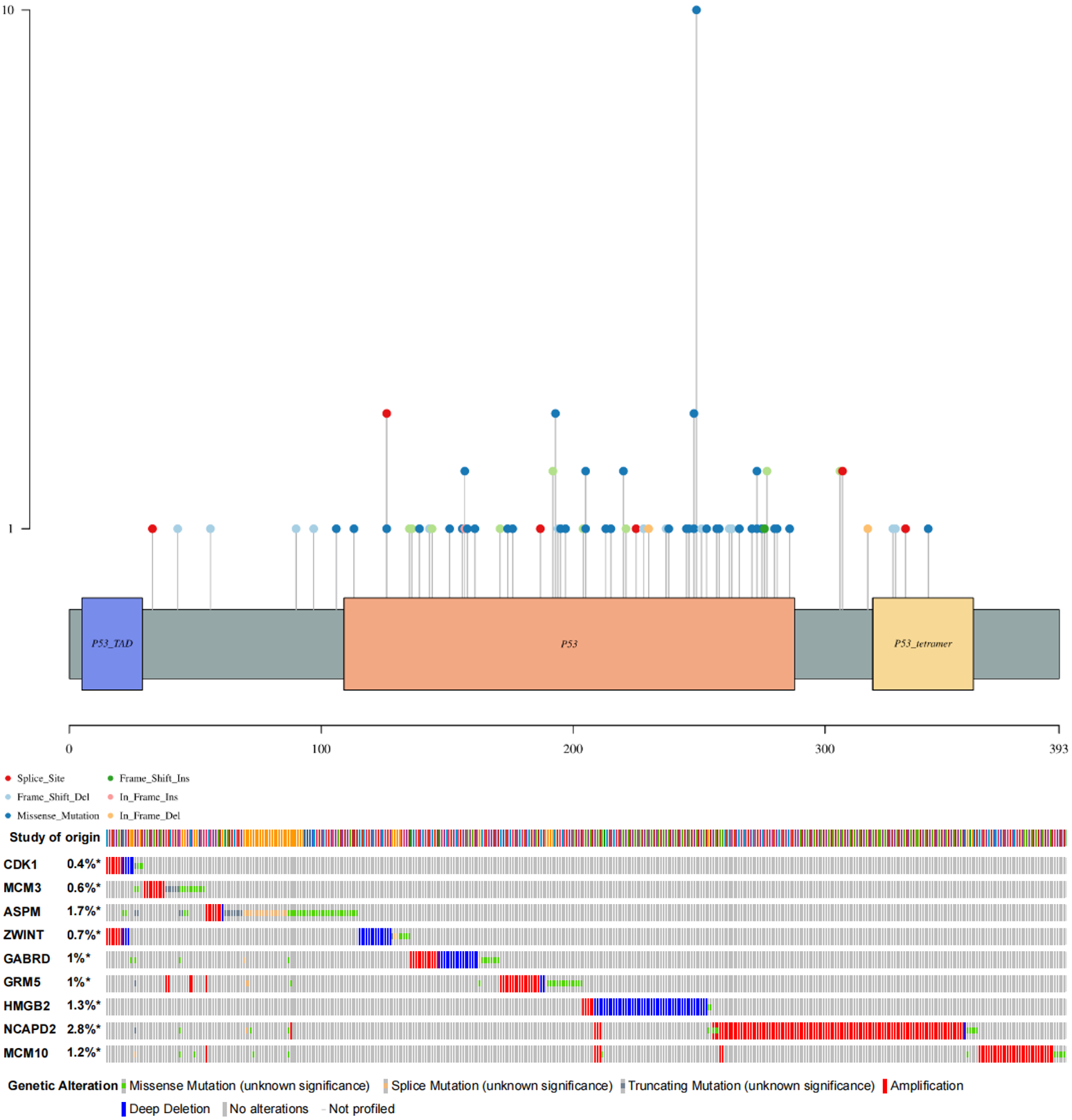
7. 基因突变情况比较
获得疾病的RNAseq数据、突变maf数据和相应的临床信息。利用R软件中的maftools软件包下载并可视化了患者的体细胞突变。
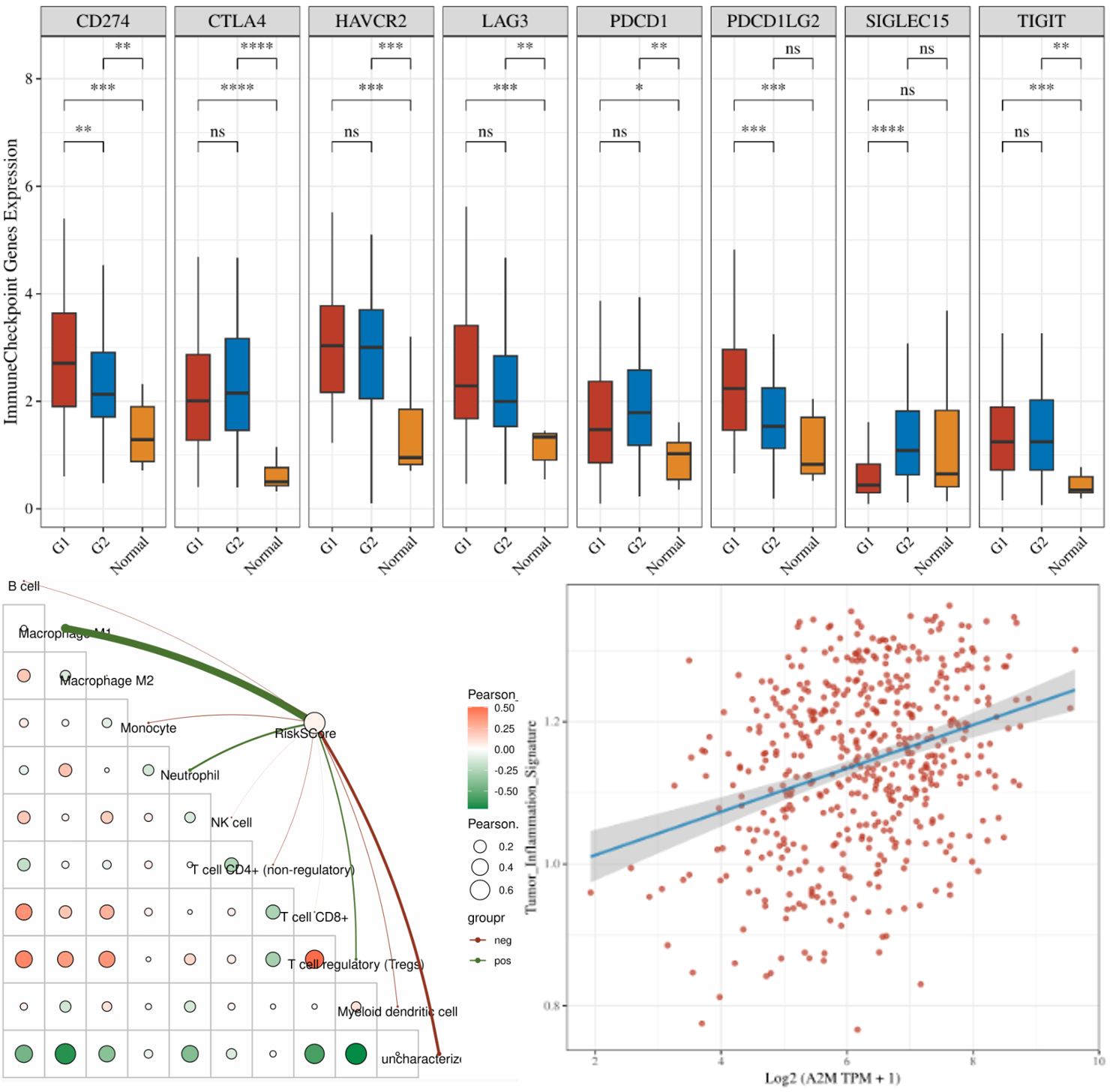
8. 免疫相关分析
简介(从数据集获得疾病的RNAseq数据和相应的临床信息。得到与与免疫检查点相关的基因,观察免疫检查点相关基因的表达情况。结果通过R软件包ggplot2和pheatmap进行实现。